Bárbara Marques1, Sofia Moeda2, Marta Contreiras2, Ana Lacerda3, Paulo Oom2.
1Department of Pediatrics, Hospital Beatriz Ângelo, Loures, Portugal and Pediatric Service, Department of Pediatrics, Hospital Santa Maria (CHLN), Lisbon Academic Medical Center, Portugal, 2Department of Pediatrics, Hospital Beatriz Ângelo, Loures, Portugal, 3Department of Child and Adolescent Oncology, Pediatric Service, Instituto Português de Oncologia Francisco Gentil, Lisbon, Portugal.
Cite this article 
Marques B, Moeda S, Contreiras M, Lacerda A, Oom P. What's hidden in the blood - A case of pancytopenia. Pediatr Oncall J. 2020;17. doi: 10.7199/ped.oncall.2020.12
|
|
| Address for Correspondence |
| Sofia Moeda, Hospital Beatriz Ângelo, Av. Carlos Teixeira, 3, 2674-514 Loures, Portugal. |
| |
| Email |
| sofia@moeda.name |
| |
| Abstract |
| A three-year-old girl presented to the emergency department with fever, cough and nasal congestion. Physical examination revealed pallor and splenomegaly. Blood analysis showed pancytopenia. She recovered quickly and uneventfully and was discharged with a diagnosis of pancytopenia caused by an unidentified virus. On outpatient follow-up, the blood counts further normalized. However, five months later, in the context of de novo facial palsy, limping and bone pain, a diagnosis of acute lymphoblastic leukemia (ALL) was made. With this case, we aim to emphasize the importance of maintaining a close follow-up of patients with pancytopenia, due to the risk of recurrence or a pre-malignant condition. |
| |
| Keywords |
| Acute lymphoblastic leukemia, child, facial palsy, pancytopenia |
| |
| Introduction |
| Acute lymphoblastic leukemia (ALL) is the most frequent malignancy in children.1-4 ALL accounts for approximately 25% of all childhood malignancies below 15 years of age and it is five times more common in children than acute myeloid leukemia (AML).2 It is more common between two and five years of age, in boys, in white children and can be associated with genetic syndromes (e.g. Down’s syndrome, Neurofibromatosis type 1, ataxia telangiectasia).1-4 Symptoms of acute leukemia are not specific and are common to other pathologies. They are due to proliferation of malignant cells in bone marrow or in other organs, which leads to changes of normal hematopoiesis and production of immature leukocytes, resulting in pancytopenia or bicytopenia with leukocytosis.2,4-6 This bone marrow aplasia may be transient in about 2% of cases of ALL in children, which is considered a pre-leukemic condition. In the majority of cases a clinical and laboratory improvement occurs, with normalization of cell counts within six months after diagnosis or in a few weeks in those who initiate corticosteroids.5-7 In this paper, we describe the case of a three-year-old girl presenting with pancytopenia of unidentified etiology that resolved spontaneously within a month. Four months later she complained of bone pain and peripheral facial nerve palsy, which led to the diagnosis of ALL. |
| |
| Case Report |
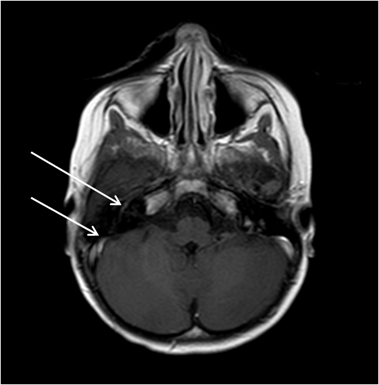
A 39 months girl with a previous history of acute pyelonephritis at 28 months of age and acute ataxia of probable viral etiology at 33 months of age presented to the emergency department with fever every four hours for four days with cough and nasal congestion. She was the second daughter of healthy non-related parents. There was no relevant family history. Physical examination revealed pallor and splenomegaly. Laboratory tests showed: hemoglobin 9.4 g/dL, mean globular volume 72 fL, leukocytes 4,700/μL (neutrophils 750/μL), platelets 61,000/μL, C-reactive protein 15.85 mg/dL, erythrocyte sedimentation rate (ESR) 57 mm at end of 1 hour, uric acid 5.3 mg/dL, lactate dehydrogenase 575 U/L, vitamin B12 510 pg/mL, folic acid 11 ng/mL. Abdominal ultrasound revealed hepatosplenomegaly. Serological tests for Epstein-Barr virus, varicella zoster virus, human herpes virus 6, human immunodeficiency virus, hepatitis A and C virus, cytomegalovirus, parvovirus B19 and leishmania were negative. Bone marrow aspirate and biopsy revealed a normocellular bone marrow, the three hematopoietic lineages having preserved morphology and maturation and no pathological infiltrates were observed. Immunophenotyping of the bone marrow showed no evidence of acute leukemia. Blood culture was negative and chest X-ray was normal. She was treated with ceftriaxone due to febrile neutropenia. During admission she recovered uneventfully; fever subsided by the fourth day and blood counts progressively normalized. She was discharged home on the ninth day with the diagnosis of pancytopenia caused by unidentified viruses. The child maintained out-patient follow-up, with regular clinical and laboratory evaluation, which remained normal. Four months later she again presented to the emergency department with right-sided peripheral facial nerve palsy. She was discharged home with a prescription for a three day course of oral prednisolone. Later the same day, she started complaining about pain and claudication of the right lower leg. Three days later, as her symptoms had not resolved, she returned to the emergency department. Physical examination showed a right-sided peripheral facial nerve palsy and pain on abduction of the right hip. Hip X-ray was normal. Laboratory analysis revealed leukocytes 17,400/μL, neutrophils 13,050/μL, C-reactive protein 2.46 mg/dL. She was discharged home with the diagnosis of transient hip synovitis and was treated with oral ibuprofen. The pain in the right lower limb resolved within three days, but two weeks later she developed pain on the feet and left wrist associated with low-grade fever (38°C). She returned to the emergency department, where on physical examination, apart from the right-sided peripheral facial nerve palsy, a discrete splenomegaly was noted. She complained of pain in lower limbs, with limitation in the mobilization of the left hip, and pain in the left upper limb. There were no inflammatory signs in the joints. A hip ultrasound failed to reveal any anomaly. The peripheral blood evaluation showed: hemoglobin 12.1 g/dL, mean globular volume 70 fL, leukocytes 66,710/μL (neutrophils 25,620/μL, lymphocytes 22,350/μL), peripheral blood smear 23% blasts, platelets 155,000/ μL, C-reactive protein 13.11 mg/dL, ESR 49 mm at end of 1 hour, uric acid 4.6 mg/dL, lactate dehydrogenase >2500 U/L, aspartate aminotransferase 187 U/L, alanine aminotransferase 18 U/L. She was started on ceftriaxone, hyperhydration and allopurinol in view of clinical suspicion of acute leukemia. She was transferred to a pediatric oncology unit, where a bone marrow aspirate and biopsy were repeated. This revealed a hypercellular bone marrow, infiltrated by 86% of lymphoblasts (Fig. 1), compatible with acute lymphoblastic leukemia (ALL). Immunophenotyping confirmed the presence of 78.8% of B lymphoblasts (CD19+/ CD79a+) with aberrant phenotype (TdT+/ CD10+/ CD34/ CD68+/ CD20+/ CD22+ weak/ cyIgM-/ IgM-), compatible with B-lineage ALL (Class II of the European Group for the Immunological Characterization of Leukemia - EGIL). Cytogenetic analysis showed the presence of the translocation t(1;19)(q23; p13) TCF3-PBX1, without rearrangements in the KMT2A gene (MLL). To evaluate central nervous system (CNS) involvement, a lumbar puncture was performed; it revealed less than five cells/mm3 and negative cytology. However, a magnetic resonance image (MRI) of the brain revealed a linear enhancement of the second and third portions of the right facial nerve, compatible with CNS infiltration (Fig 2). The final diagnosis was of a high-risk B-lineage ALL and the patient started treatment according to the therapeutic protocol DFCI-ALL 2011 (Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia).
Figure 1 - Myelogram (hematoxylin-eosin): pancytopenia (1) and lymphoblasts infiltration (2).
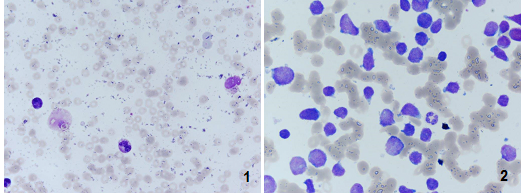
Figure 2 - Magnetic resonance imaging of the brain (T1 with gadolinium): linear enhancement in topography to the second and third portions of the right facial nerve.
|
| |
| Discussion |
Frequently, the initial suspicion of ALL arises from an evaluation of a complete blood count and peripheral blood smear. These commonly show cytopenias, although absolute leukocyte count may be increased due to circulating blasts. This hypothesis must be confirmed by a bone marrow aspirate with morphological, immunophenotype, molecular and genetic studies, to fully describe the disease and define prognosis. When this is inconclusive or if marked pancytopenia is present, it is also recommended to perform a bone marrow biopsy to increase the likelihood of obtaining abnormal cells.2,3,8 Despite an initial normal bone marrow aspirate and biopsy in our patient, a malignancy was later diagnosed. In fact, several authors describe a pre-leukemic stage before a definitive diagnosis of ALL, more common in girls younger than ten years of age and usually associated with signs of viral or bacterial infection. At this stage, the patient presents with pancytopenia and normal or hypocellular bone marrow, suggestive of bone marrow aplasia. This is a transient finding, resolving within six months without specific treatment or in only a few weeks when steroid treatment is started.5-7,9,11 The etiologic relationship between initial bone marrow aplasia and subsequent diagnosis of an ALL is not yet well established. Several theories are advocated, such as existence of a prior malignant clonal population (true pre-leukemic state), or of an immunosurveillance mechanism temporarily controlling the pre-leukemic burden, occurrence of a viral or bacterial infection leading to the suppression of hematopoiesis, production of endogenous corticosteroids secondary to infection with elimination of lymphoblasts or exposure to exogenous toxins.5,6,9,11
Cytopenias are a consequence of replacement of normal bone marrow by malignant hematopoietic cells, which generally proliferate as a monoclonal population and infiltrate other organs (spleen, liver, lymph nodes, central nervous system).1,4 Thus, patients with ALL have an increased risk of involvement of the CNS, with cranial neuropathies, meningeal signs and symptoms of increased intracranial pressure (headache, vomiting, alterations in conscious state, ocular changes).2,3,8 In our patient, peripheral facial nerve palsy was present before ALL diagnosis and treatment with steroids was initiated. Although there are no definitive recommendations for the use of corticosteroids in peripheral facial paralysis, many pediatricians choose to institute therapy, as it is assumed that this will decrease inflammation and edema of the facial nerve sheath. However, in the case of ALL, it may delay the diagnosis and compromise the prognosis10. The presence of bone pain simultaneously with the peripheral facial nerve palsy should enhance the suspicion of ALL, since musculoskeletal pain may be a presenting symptom in more than 40% of cases of ALL.2 It is also important to note that the presence of peripheral facial nerve palsy in a child with ALL diagnosis should lead to suspicion of a possible CNS infiltration, which was confirmed in this case by MRI of the brain, although the cerebrospinal fluid cytochemical examination was normal.
In conclusion, we consider it is essential to maintain a regular out-patient follow-up of cases of resolved pancytopenia without a known cause, due to the chance of recurrence and/or a malignant etiology. Furthermore, the presence of bone pain or facial nerve palsy should draw attention to the risk of leukemia. In a patient with an ALL diagnosis, the possibility of CNS involvement (which can be represented, for instance, by facial nerve palsy) should be kept in mind. |
| |
| Funding |
| None |
| |
| Conflict of Interest |
| None |
| |
| References : |
- Weinzierl EP, Arber DA. The differential diagnosis and bone marrow evaluation of new-onset pancytopenia. Am J Clin Pathol 2013; 139: 9-29 [CrossRef] [PubMed]
|
|
No comments:
Post a Comment