Mukesh Sanklecha, Nayan Chaudhary, Chinmay Kumbhar.
Department of Pediatrics and Pediatric Intense Care Unit, Bombay Hospital Institute of Medical Sciences, Mumbai, India.
Cite this article 
Sanklecha M, Chaudhary N, Kumbhar C. Recurrent Respiratory Distress - Is It Microscopic Polyangiitis?. Pediatr Oncall J. 2019;16. doi: 10.7199/ped.oncall.2019.59
|
|
| Address for Correspondence |
| Dr. Mukesh Sanklecha, Bombay Hospital Institute of Medical Sciences, 12, New Marine Lines, Mumbai 400020, India. |
| |
| Email |
| doctormukesh@gmail.com |
| |
| Keywords |
| Microscopic polyangiitis, ANCA vasculitis, crescentic glomerulonephritis |
| |
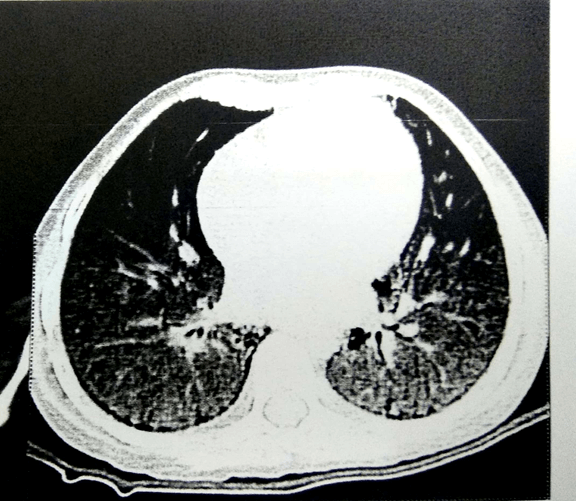
A 4½ years old boy presented with respiratory distress and fever for 15 days duration. He had similar episodes of fever and respiratory distress 3 times previously in the past 2½ years each time needing hospitalization, supplemental oxygen, intravenous antibiotics and prolonged pediatric intensive care unit (PICU) stay. On presentation, he was malnourished (weight 11 kg and height 111 cms, both below the 3rd centile), had clubbing, pallor, mild hepatosplenomegaly and moderate respiratory distress (respiratory rate of 40/min) with heart rate of 120/min and oxygen saturation of 88 percent at room air. Investigations showed hemoglobin 9.4 gm/dl, white blood count 18,000 cells/cumm with 69% polymorphs, ESR of 39 mm at end of 1 hour. Serum creatinine was 1 mg/dl and liver function tests were normal. Chest CT scan revealed bilateral parenchymal consolidation and nodular air space opacities with diffuse ground glass haziness in both lungs with multiple sub-pleural cysts and mild mediastinal and hilar lymphadenopathy suggestive of interstitial lung disease (Figure 1). Bronchoalveolar lavage (BAL) did not grow any organism on bacterial, fungal culture or TB culture. GeneXpert on BAL was negative. Hemoglobin electrophoresis, bone marrow aspirate were normal. CFTR gene test was negative. HIV, nitroblue tetrazolium test were negative and serum immunoglobulin levels and lymphocyte subsets on flow cytometry were normal. ANA was weakly positive with a titre of 1:80, dsDNA was negative. Urine examination was normal. He was treated for this episode with intravenous ceftriaxone and oral azithromycin and was discharged after three weeks. Three months later he was re-hospitalized for further evaluation as a urine examination in between had revealed microscopic hematuria with 30-50 red blood cells (RBCs) per high power field (hpf) with a normal creatinine and no growth on a urine culture. Kidney biopsy revealed crescentic glomerulonephritis (fibrocellular/fibrous crescents detected in 8 out of 16 glomeruli) and an immune work up revealed an MPO-ANCA titre of 123.94 RU/ml (normal < 20) with a negative PR3-ANCA. He was diagnosed to have ANCA vasculitis and crescentic nephritis along with pulmonary involvement and was treated with rituximab (750mg/m2/dose IV for two doses two weeks apart)1 along with pulse intravenous methylprednisolone (30 mg/kg /dose once a day for 3 days) followed by cyclophosphamide 2.5 mg/kg/day for 3 months along with a baseline low dose steroid cover. However, due to non-response, cyclophosphamide was shifted to oral azathioprine (1.5 mg/kg/day once daily). On follow-up over 2 years, the respiratory symptoms have never recurred and the hematuria also settled. However, there is no increase in weight. His renal functions remain stable.
Figure 1. CT scan of the chest shows diffuse ground glass haziness in both lungs with multiple sub-pleural cysts
The anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitides (AAV) are a group of diseases with necrotizing inflammation that affects the small and medium sized vessels with autoantibodies against the cytoplasmic region of the neutrophil.2 Included under this umbrella are granulomatosis with polyangiitis (GPA formerly known as Wegener’s granulomatosis), microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (formerly known as Churgh Strauss syndrome).2 A diagnosis of GPA can be made when at least three of the following six criteria are met: a) granulomatous inflammation on histopathology, b) upper airway involvement, c) upper laryngo-tracheo-bronchial stenosis, d) pulmonary involvement, e) ANCA positivity and f) renal involvement.1,3 If there is no clinical evidence of granulomatous inflammation or eosinophilic vasculitis, the AAV syndrome is termed as MPA.2 Since our child had pulmonary involvement, renal involvement and ANCA positivity with no granulomatous inflammation or eosinophilic vasculitis, a diagnosis of MPA was considered. Non-specific constitutional symptoms actually represent the most common presenting complaint leading to a significant delay in diagnosis in most cases.3,4,5 That probably explains the repeated hospitalizations, extensive workup and the time interval between the patient presenting to us and the final diagnosis. Traditionally cyclophosphamide is commonly used as the initial drug for induction. However recent reports suggest that rituximab may be equally effective and especially useful in severe or life threatening disease as first line therapy.1,6,7 We used rituximab for initial induction followed by cyclophosphamide when rituximab gave no results. Maintenance therapy was continued with azathioprine.3 While the prognosis of this condition is clearly improving with newer evolving therapies, the risk for progression to end stage renal disease remains, as does the risk of toxicity due to the potent drugs involved. |
|
No comments:
Post a Comment