Nightly Fever, Swollen Lymph Nodes, and Rash in a 3-Year-Old
The Case Challenge series includes difficult-to-diagnose conditions, some of which are not frequently encountered by most clinicians but are nonetheless important to accurately recognize. Test your diagnostic and treatment skills using the following patient scenario and corresponding questions. If you have a case that you would like to suggest for a future Case Challenge, please contact us.
Background
A 3-year-old boy with no significant past medical history presented to the emergency department (ED) with 10 days of nightly fever. He also had cervical lymphadenopathy and an intermittent erythematous macular rash similar to the one shown below.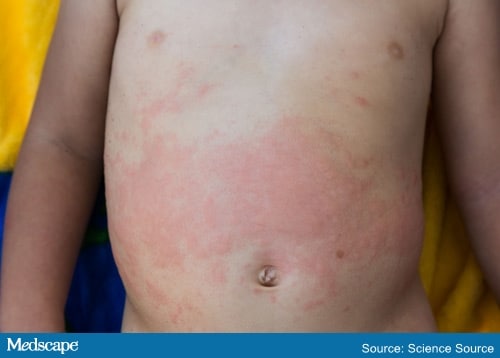 His parents also stated that he exhibited pain bilaterally in his knees, ankles, and hips. Before the onset of the fevers, he had been well. His parents reported no other symptoms, including conjunctivitis, oral mucosal changes, hand or foot swelling, rhinorrhea, cough, dyspnea, abdominal pain, vomiting, or diarrhea. He had no known sick contacts. His parents reported no travel outside the United States. The family has no pets. He has one older brother, who is healthy. The parents reported no relevant family history.
His parents also stated that he exhibited pain bilaterally in his knees, ankles, and hips. Before the onset of the fevers, he had been well. His parents reported no other symptoms, including conjunctivitis, oral mucosal changes, hand or foot swelling, rhinorrhea, cough, dyspnea, abdominal pain, vomiting, or diarrhea. He had no known sick contacts. His parents reported no travel outside the United States. The family has no pets. He has one older brother, who is healthy. The parents reported no relevant family history.Physical Examination and Workup
Upon examination, the patient was febrile and irritable, with large, palpable cervical lymph nodes. He had swelling of his bilateral knees and ankles and was reluctant to bear weight. His abdomen was diffusely tender to palpation, and he had mild hepatosplenomegaly. He had an erythematous macular rash, which displayed the Koebner phenomenon.On the basis of his history, as well as physical examination and laboratory findings, the patient was diagnosed with incomplete Kawasaki disease (KD) and admitted for treatment. He received standard therapy with intravenous immunoglobulin (IVIG) at 2 g/kg and aspirin. However, more than 36 hours after treatment, he still had nightly fevers. He was treated with another dose of IVIG. Although he remained febrile, his laboratory findings appeared to improve. His white blood cell count, platelet count, and ESR all began to decrease. However, his cell counts did not stabilize once they reached normal levels. Within 24 hours, the patient became leukopenic, thrombocytopenic, and more anemic than he was at initial presentation. His transaminitis worsened. Further laboratory workup revealed a low fibrinogen level, prolonged prothrombin time, and partial thromboplastin time, and a ferritin level > 10,000 ng/L. The patient became lethargic, with worsening rash and unremitting fever.
Discussion
Macrophage activation syndrome (MAS) is a severe and potentially fatal immune phenomenon characterized by overactivation and proliferation of macrophages and T cells. It is closely related to hemophagocytic lymphohistiocytosis (HLH), a similar diagnosis that may occur as a result of genetic factors or certain malignancies. HLH and MAS are thought to exist along a spectrum, and MAS has been described as a type of secondary HLH that occurs in patients with an underlying rheumatologic disorder. Although most closely associated with systemic-onset juvenile idiopathic arthritis (SoJIA), MAS is also associated with numerous other rheumatologic diseases, including systemic lupus erythematosus and KD.- Platelet count ≤ 181 × 109/L
- AST level > 48 U/L
- Triglyceride level > 156 mg/dL
- Fibrinogen level ≤ 360 mg/dL
Question 1 of 2
MAS is a severe and potentially fatal immune phenomenon that can occur as a complication of numerous rheumatologic diseases, most prominently SoJIA. It is closely related to HLH; however, HLH occurs because of genetic factors or certain malignancies, whereas MAS occurs secondary to rheumatologic disease. MAS may occur at any time in a patient with SoJIA and may be triggered by treatment for the disease. For a definitive diagnosis of SoJIA, patients must have fevers for 2 weeks and arthritis for 6 weeks; however, in patients with characteristic symptoms, particularly those with MAS on presentation, treatment can (and should) be initiated before this time.
Question 2 of 2
Criteria for MAS complicating SoJIA state that a febrile patient with known or suspected SoJIA is classified as having MAS if they have a ferritin level ≥ 684 ng/L, in addition to two of the following:
- Platelet count ≤ 181 × 109/L
- AST level > 48 U/L
- Triglyceride level > 156 mg/dL
- Fibrinogen level ≤ 360 mg/dL
References
- Minoia F, Davì S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66:3160. Source
- Ravelli A, Minoia F, Davì S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2016;75:481-489. Source
- Minoia F, Bovis F, Davì S, et al. Development and initial validation of the macrophage activation syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that differentiates primarily hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr. 2017;189:72. Source
- Rajasekaran S, Kruse K, Kovey K, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children. Pediatr Crit Car Med. 2014;15:401-408. Source
Cite this: Sarah Taber. Nightly Fever, Swollen Lymph Nodes, and Rash in a 3-Year-Old - Medscape - Dec 16, 2019.
No comments:
Post a Comment